Protein folding per tutti
In due articoli pubblicati giovedì su Nature e Science, la società londinese DeepMind specializzata in tecniche di deep learning e un gruppo di ricercatori guidato da David Baker, biologo strutturale della University of Washington, hanno descritto due algoritmi basati su reti neurali profonde che prevedono in modo estremamente accurato la struttura delle proteine a partire dalle sequenze di aminoacidi. In alcuni casi la loro precisione è confrontabile con quella delle strutture misurate sperimentalmente. Contestualmente hanno messo a disposizione gratuitamente il codice per il calcolo di queste strutture.
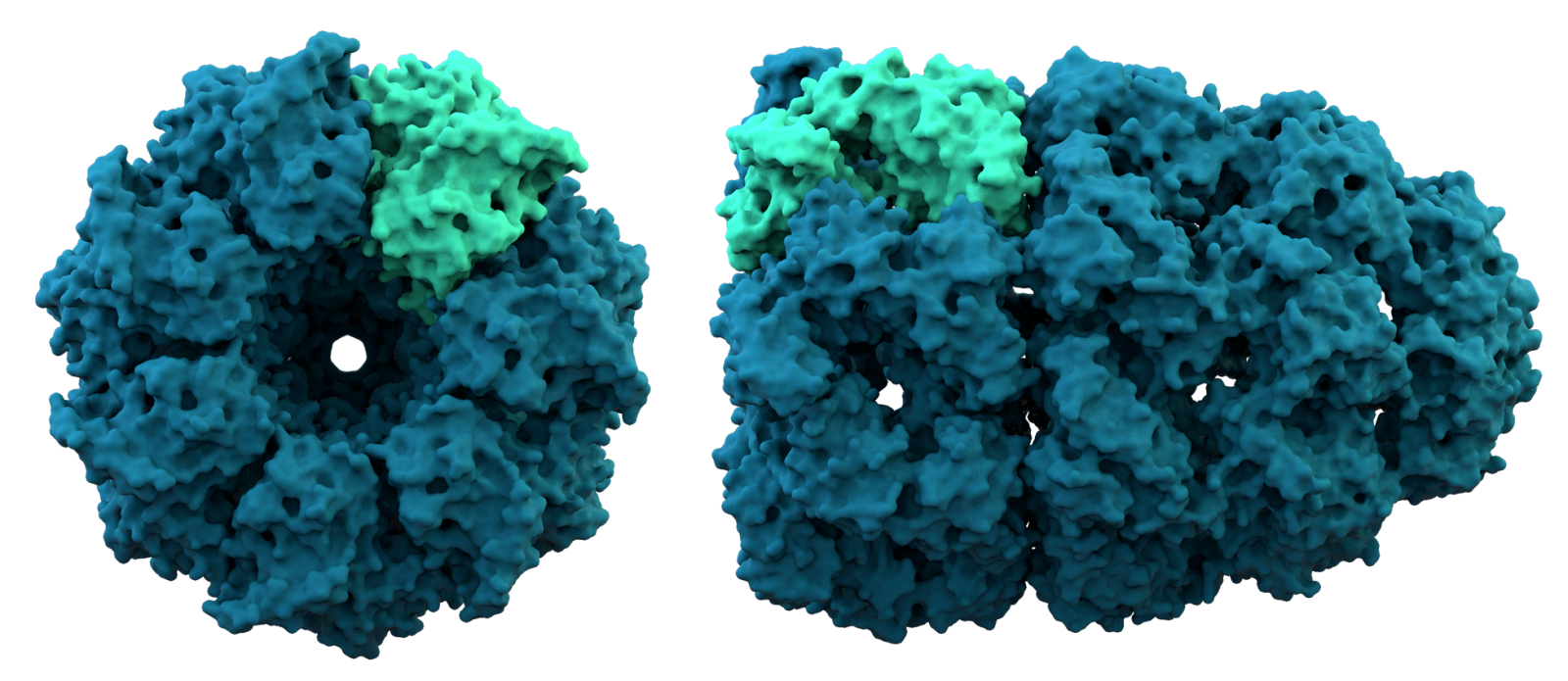
Nell'immagine: struttura cristallina del complesso proteico chaperonina. Credit: Thomas Splettstoesser/Wikimedia Commons. Licenza: CC BY-SA 3.0.
Le proteine sono le molecole fondamentali per i processi biologici e la loro struttura tridimensionale, cioè il modo in cui gli aminoacidi che le compongono sono distribuiti nello spazio, è strettamente legata alle funzioni che svolgono. Conoscere questa struttura è un compito tutt’altro che semplice. Sperimentalmente può essere estremamente oneroso e in alcuni casi impossibile. Da cinquant’anni gli scienziati cercano di sviluppare dei metodi computazionali, che inferiscano la struttura a partire dalla sequenza di aminoacidi che costituiscono la proteina.








{kind=link}
{kind=link}
.jpg){kind=link}