
Prima la Gran Bretagna, poi il Sudafrica e da ultimo il Brasile: le nuove “versioni” del coronavirus sono al centro dell’attenzione degli scienziati e dell’opinione pubblica. Cosa ne sappiamo? Sono più pericolose? Possono davvero cambiare la traiettoria della pandemia e mettere a rischio l’efficacia dei vaccini e delle terapie? Perché è importante sequenziare il virus?
Durante i mille anni che separano la caduta dell’Impero Romano d’Occidente dall’invenzione della stampa, nei monasteri benedettini sparsi per l’Europa legioni di monaci amanuensi copiavano su robusti fogli di pergamena ricavati dalla pelle di agnello o di vitello i testi antichi, tramandati su fragili rotoli di papiro e sopravvissuti al trascorrere del tempo e alle vicissitudini politiche e militari dell’Alto Medioevo. È a questi eroi, in gran parte sconosciuti, che dobbiamo riconoscenza per averci trasmesso il patrimonio di classici greci e latini che ancora oggi costituiscono la base della nostra cultura.
Durante questa attività i monaci, com’è umano, commettevano errori di copiatura: alcuni casuali, altri dovuti all’imperfetta conoscenza del greco e dell’arabo da cui spesso traducevano in latino, altre volte infine perché omettevano di copiare pagine o interi volumi ritenuti immorali o sconvenienti per gli standard morali e religiosi dell’epoca. Il celebre romanzo di Umberto Eco Il Nome della Rosa è ambientato appunto in un monastero di amanuensi benedettini, e la sua trama si sviluppa intorno a uno di questi testi proibiti, l’immaginario secondo volume della Poetica di Aristotele dedicato al riso e alla commedia.
Filologia e filogenetica
Sono sorprendenti le analogie tra l’attività dei filologi, che comparano le varie copie di un testo antico per risalire, dal confronto tra gli errori di copiatura, al testo originale così come era stato redatto dall’autore, e il lavoro che fanno oggi gli studiosi di filogenetica dei virus, i quali mettono a confronto le varie “versioni” di un virus e, confrontando gli errori di replicazione, riescono a risalire all’antenato comune e alla data dello spillover, il momento in cui il virus – nel nostro caso il SARS-CoV-2 - è saltato per la prima volta dal suo reservoir naturale (quasi certamente un pipistrello) all’essere umano, per il tramite di un ospite intermedio ancora sconosciuto (furetto? pangolino?).
Le analogie non finiscono qui. Una recente ricerca1, realizzata da matematici e bio-informatici del MIT di Boston e recentemente pubblicata su Science, ha applicato alle mutazioni delle proteine virali le tecnologie di machine learning utilizzate per lo studio dei linguaggi naturali e della loro evoluzione. Ci torneremo più avanti. Per poter leggere il codice del virus occorre effettuare un'operazione detta sequenziamento genomico, con la quale, a partire da un campione biologico, si ottiene una trascrizione del genoma, cioè l’ordine esatto nel quale si dispongono, lungo i due filamenti che formano la “doppia elica” del DNA, le quattro basi azotate (adenina, citosina, guanina e timina) che costituiscono l’alfabeto della vita. Nel caso del nostro virus, il cui genoma è costituito invece da un singolo filamento di RNA, troviamo un’altra base, l’uracile, al posto della timina.
Quando il virus entra nel citoplasma delle cellule umane, utilizzando la proteina spike come una chiave falsa per forzare i recettori ACE2 presenti sulla superfice delle cellule, prende possesso dei meccanismi di produzione delle nuove molecole della cellula, che inizia così a produrre tante copie del RNA del virus anziché il proprio. Ma la natura non è perfetta e così, in questo processo di replicazione all’interno delle cellule umane, il virus può “mutare”, possono cioè verificarsi errori casuali nel processo di trascrizione del genoma virale, e le nuove copie di RNA possono avere modifiche rispetto a quella originaria.
Il caso e Darwin
Il ruolo dei filogenetisti non è quindi soltanto quello di guardare indietro e cercare di ricostruire la storia passata del virus, ma anche quello di osservare il percorso evolutivo del patogeno e le sue mutazioni, attuali e potenziali. Dal momento che questo processo avviene continuamente, l’emergere di nuove varianti è un evento prevedibile, non è di per sé motivo di preoccupazione, e il virus SARS-CoV-2 non fa eccezione. Ci sono virus più o meno soggetti a mutare, ma le evidenze che emergono dai primi mesi della pandemia confermano il SARS-CoV-2 come un virus abbastanza stabile.
In una ricerca pubblicata su Nature2 alla fine di novembre 2020 sono stati analizzati oltre 46.000 genomi virali sequenziati in tutto il mondo, e non sono state identificate in questo insieme mutazioni ricorrenti associabili in modo convincente a una maggiore trasmissione. Presupponendo infatti che la frequenza degli errori sia costante, nel caso del SARS-CoV-2 è valutata in un errore ogni 10.000 caratteri trascritti. L’abbondanza degli errori dipende solo dalla lunghezza del genoma (ovvero del “testo” da trascrivere) e dalla presenza o meno di sistemi di correzione degli errori: qualcosa di simile a ciò che fanno i correttori di bozze nelle redazioni giornalistiche. In effetti il SARS-CoV-2, che è uno dei virus a RNA con il patrimonio genetico più grande, ha anche un meccanismo di correzione degli errori che mitiga gli effetti degli errori di trascrizione, riducendone il numero. Alcuni errori sfuggono comunque alla correzione e va detto che si tratta, comunque, di errori casuali.
La maggior parte delle mutazioni che ne derivano non ha un impatto significativo sulla diffusione del virus, ma alcune mutazioni o combinazioni di mutazioni possono fornire al virus “variato” un vantaggio evolutivo, come una maggiore trasmissibilità o la capacità di eludere la risposta immunitaria dell’ospite. Darwin ci ha insegnato che il più forte, quello che si adatta meglio, sopravvive; se quindi una mutazione del genoma, generatasi casualmente, permette al virus di riprodursi con maggiore efficienza e rapidità e/o di adattarsi meglio all’ambiente nel quale si trova, può diventare dominante e soppiantare il ceppo originario o altre varianti meno efficienti dal punto di vista evolutivo.
La fabbrica delle proteine
Facciamo un passo indietro e torniamo all’interno della nostra cellula infetta, dove il virus ha creato tante copie del proprio genoma. L’RNA virale, in sé, non è ancora un nuovo virus, è piuttosto un “libretto di istruzioni”, una sequenza di comandi per la produzione delle varie proteine che poi si assembleranno, come i mattoncini del Lego, a formare nuove copie del virus. Queste “fabbriche delle proteine” si chiamano ribosomi, e sono anch’esse contenute nel citoplasma delle cellule. All’interno del ribosoma si trova una molecola, il tRNA, che legge la sequenza delle circa 30.000 basi azotate (o nucleotidi) allineate lungo il filamento di RNA del SARS-CoV-2, e sulla base delle istruzioni in esse contenute, sintetizza gli amminoacidi che vanno a costituire le proteine del virus.
Le basi vengono lette a gruppi di tre, quindi ogni tripletta (detta anche codone) può avere 64 combinazioni diverse: a 61 di esse corrisponde uno dei 20 amminoacidi necessari alla creazione delle proteine, mentre le restanti tre combinazioni sono utilizzate come segnale di stop, ovvero indicano che la “parola” da leggere è stata completata e che la prossima lettura sarà l’inizio di una parola diversa, esattamente come gli spazi vuoti in un testo scritto.
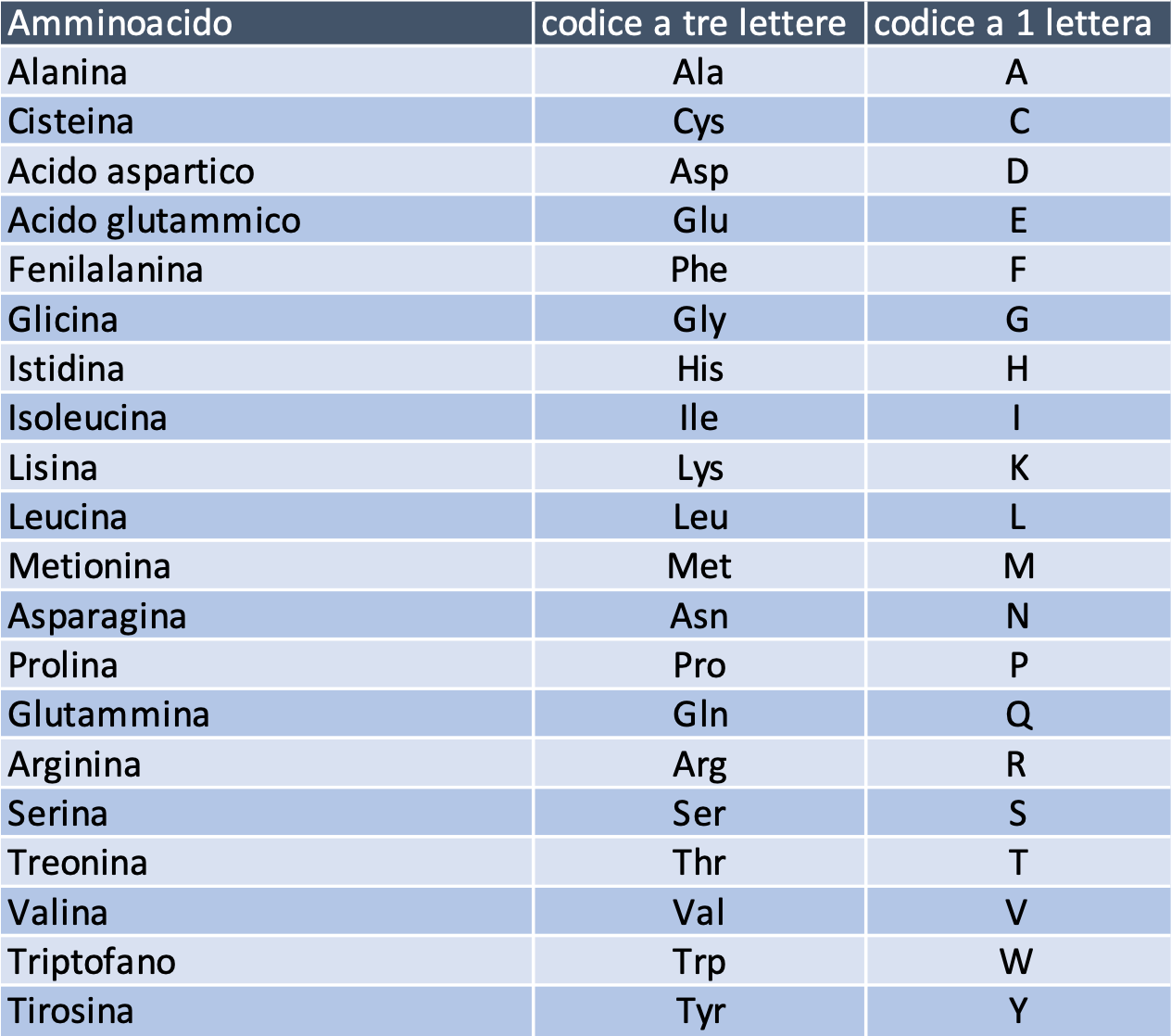
Figura 1. I 20 aminoacidi ordinari
Gli errori di replicazione del RNA virale possono portare a una diversa collocazione di alcune delle basi lungo il filamento del RNA, alla loro cancellazione, o all’inserimento di nuove basi. Quando leggiamo per esempio che la variante inglese comprende la mutazione N501Y, vuol dire che a seguito di una diversa composizione delle basi in essa contenute, la tripletta posta nella posizione 501 di quella proteina ha prodotto, anziché l’asparagina (contrassegnata col codice N), la tirosina (codice Y). La modifica può non avere alcun effetto, se per esempio l’amminoacido prodotto dalla tripletta modificata è lo stesso di quella originaria (ricordiamo: abbiamo 61 possibili triplette per produrre 20 amminoacidi): in questo caso si parla di “mutazione silente” o “sinonima”; ma può anche portare alla produzione di un amminoacido al posto di un altro (“scambio”), alla perdita totale di uno o più amminoacidi (definita dai genetisti “delezione”, perché comporta il taglio di una parte della proteina), all’inserimento di amminoacidi non presenti nel genoma originale, alla troncatura della parte rimanente di una proteina, che si verifica quando una tripletta che codifica un amminoacido viene cambiata con una che codifica il segnale di stop (“non senso”), o viceversa alla cancellazione della codifica di stop con una che codifica un amminoacido, col risultato di creare una proteina più grande di quella originale e probabilmente disfunzionale (“no stop”).
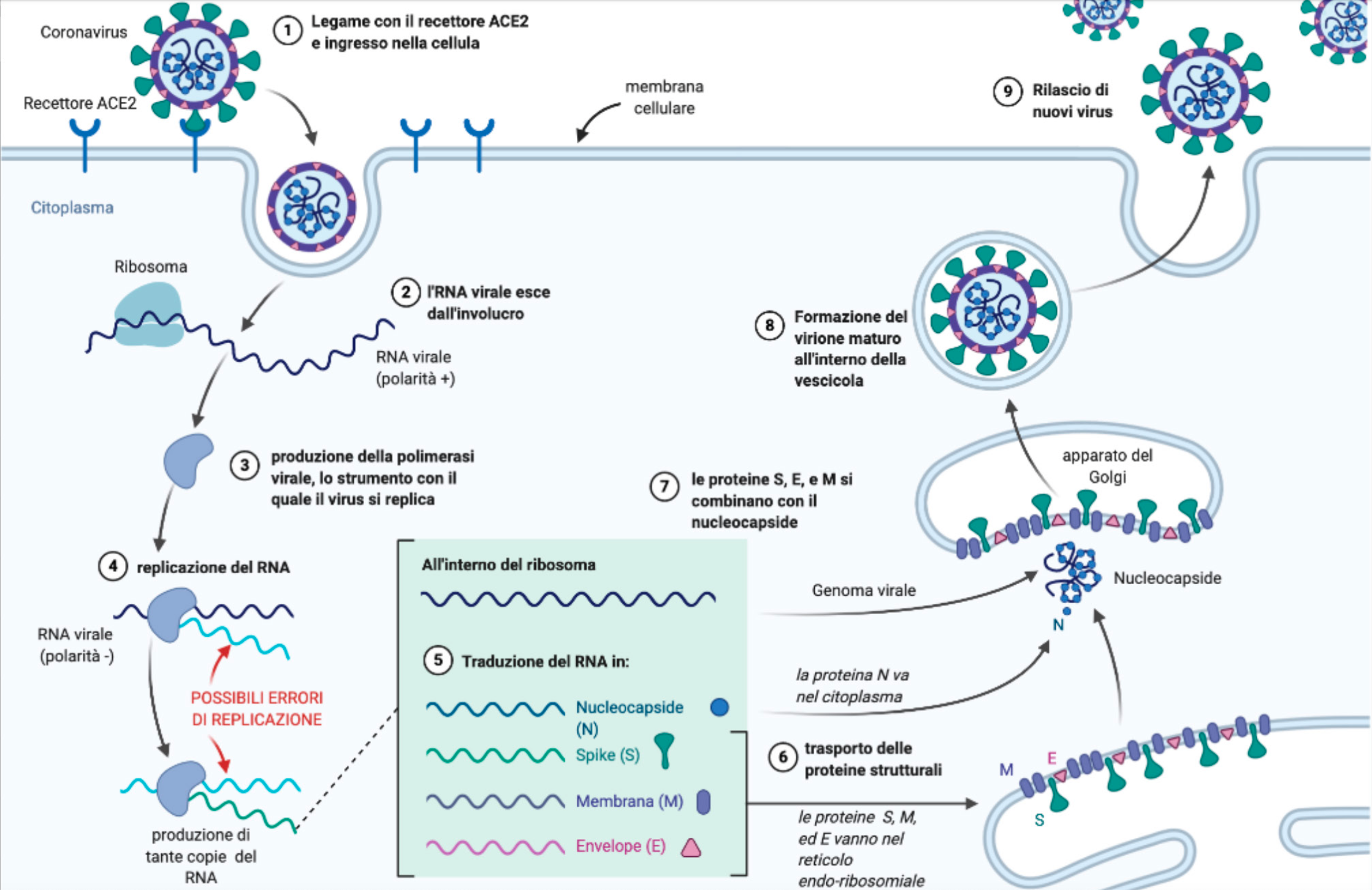
Figura 2. Il ciclo di replicazione di SARS-CoV-2 all'interno delle cellule umane. Immagine creata da Tommaso Curiale con BioRender
Il virus “reagisce” all’uomo?
L’infezione da SARS-CoV-2 ha generalmente una durata di una settimana-dieci giorni. Dopo questo periodo, a parte i casi in cui, soprattutto nelle persone più anziane o che sono affette anche da altre patologie di rilievo, l’infezione assume forme gravi o critiche e progredisce verso la polmonite interstiziale, l’infezione viene risolta grazie agli anticorpi prodotti dal sistema immunitario umano, che neutralizzano le proteine del virus e ne bloccano l’entrata nelle cellule. A quel punto il virus, essendo un parassita obbligato che alla lunga può sopravvivere soltanto all’interno di altri esseri viventi, deve riuscire a contagiare un altro ospite suscettibile all’infezione, nel nostro caso un altro essere umano, oppure giunge a fine corsa.
Questo virus è entrato in contatto con l’uomo solo da poco tempo, e il nostro sistema immunitario è sicuramente più aggressivo nei suoi confronti rispetto a quello degli animali che costituiscono il suo serbatoio, quasi certamente i pipistrelli, nei quali ha probabilmente trovato un equilibrio. Per dirla in maniera un po’ semplicistica, le due parti hanno trovato un accordo: il virus non uccide chi lo ospita e cerca di non danneggiarlo, e l’animale ospite gli consente di sopravvivere e moltiplicarsi a lungo pressoché indisturbato.
Il virus, specie quando “nuovo”, è dunque sottoposto a una forte pressione selettiva, che riguarda soprattutto le proteine più esposte all’attacco del sistema immunitario, e tra queste la proteina spike, che esercita un ruolo fondamentale nel consentire l’infezione delle cellule. La proteina spike è infatti il bersaglio principale degli anticorpi neutralizzanti, di quelli prodotti a seguito di infezione “naturale” ma anche di quelli la cui produzione è stimolata dai vaccini, che di fatto “simulano” una infezione naturale introducendo nel corpo umano proprio questa proteina “preformata” oppure le istruzioni per farla produrre da parte delle stesse cellule umane.
Se dunque le mutazioni in altre porzioni del genoma virale possono avere poca o nessuna importanza evolutiva, nel caso della proteina S è più probabile che prendano il sopravvento e si affermino le mutazioni che danno al virus qualche chance in più di resistere all’attacco degli anticorpi. Alcuni ricercatori hanno avanzato l’ipotesi che l’affermarsi di varianti come quella inglese possano essere state accelerate dalla prolungata replicazione del virus nell’organismo di pazienti immunocompromessi che spesso manifestano un’infezione di lunga durata3. In tal caso il virus, meno “disturbato” dagli anticorpi umani, avrebbe agio di replicarsi più a lungo all’interno dell’ospite e di adattarsi meglio. Un recente studio4 ha analizzato il caso di un paziente oncologico trattato con un farmaco che riduce la produzione di linfociti B, deceduto 101 giorni dopo aver contratto l’infezione. Per i primi due mesi dopo l’infezione il virus si è replicato senza significative mutazioni, ma a seguito di un ciclo di trattamento con plasma di convalescente il virus ha sviluppato significative mutazioni.
In una corrispondenza alla rivista New England Journal of Medicine5 è documentato un caso simile: un paziente immunocompromesso deceduto dopo 154 giorni dall’infezione, che durante il decorso clinico è stato trattato tra l’altro con corticosteroidi e altri farmaci presumibilmente in grado di esercitare una pressione selettiva, quali idrossiclorochina (farmaco antimalarico con effetto anche antinfiammatorio), remdesivir (inibitore della replicazione dei virus a RNA), immunoglobuline per endovena, e un cocktail sperimentale di anticorpi monoclonali. L’analisi filogenetica dei campioni prelevati al paziente durante la sua storia clinica ha evidenziato un'accelerazione nell’evoluzione del virus, con la maggior parte delle mutazioni intervenute nella proteina Spike.
In una terza case history6, riguardante una paziente oncologica sopravvissuta a un'infezione durata 105 giorni, il sequenziamento effettuato su un campione virale prelevato 70 giorni dopo l’inizio della malattia ha evidenziato la presenza di numerose variazioni genetiche rispetto a un altro sequenziamento fatto al giorno 49, prima che il paziente ricevesse due infusioni di plasma di convalescente. E potrebbe non essere un caso che una delle varianti più importanti di cui sinora si ha notizia sia emersa in un paese, il Sudafrica, che ha una delle maggiori percentuali al mondo di persone sieropositive all’HIV, quindi con un sistema immunitario indebolito.
I tre imputati
Al momento, le varianti che preoccupano maggiormente in un’ottica di contenimento della pandemia sono tre, e in base alla nazione nella quale sono emerse sono state rispettivamente ribattezzate “inglese”, “sudafricana” e “brasiliana”, anche se l’Oms sta valutando l’adozione di un sistema unico di denominazione, per semplificare la nomenclatura tecnica e soprattutto per evitare l’associazione di un patogeno o di una sua variante a specifiche aree geografiche: l’esperienza del “virus cinese” docet.
La più nota di queste varianti è quella che si è diffusa a partire dalla fine del mese di settembre nel sud-est della Gran Bretagna e la cui denominazione tecnica è VOC-202012/01 (letteralmente “variante oggetto di attenzione n.1 del dicembre 2020”) o B.1.1.7. Questa variante ha causato e continua a causare grande allarme, e indotto il governo inglese ad assumere significative misure di contenimento, dapprima nel sud-est e a Londra, dove rappresenta ormai oltre il 50% dei nuovi casi, e successivamente in tutto il paese, dove nello stesso periodo l’incidenza dei casi positivi è aumentata in maniera significativa7. Secondo due analisi, elaborate rispettivamente dalla London School of Hygyene and Tropical Medicine8 e dall’Imperial College di Londra9, questa nuova variante sarebbe caratterizzata da una maggiore trasmissibilità rispetto agli altri ceppi più comuni.
La variante B.1.1.7 presenta diverse e significative modifiche del genoma rispetto al ceppo originario; quelle più rilevanti ai fini dell’evoluzione della pandemia sono le otto che riguardano la proteina spike, e in particolare la HV 69-70 deletion, ovvero la perdita degli amminoacidi nelle posizioni 69 e 70, che potrebbe causare una diversa conformazione della proteina, mutandone l’aspetto, e la mutazione N501Y, che trovandosi in quell’area della proteina Spike detta RBD (Receptor Binding Domain), che si lega ai recettori della cellula umana ACE2, potrebbe renderla meno attaccabile dagli anticorpi che sono stati sviluppati contro un virus che non possedeva questa mutazione, quale quello che finora è circolato e contro il quale sono stati prodotti anticorpi e vaccini. Un tipo di test diagnostico molecolare molto utilizzato nel Regno Unito non riesce a rilevare la proteina spike quando presenta questa delezione: esso viene pertanto utilizzato per diagnosticare la positività alla variante inglese, quando il test restituisce un valore negativo alla proteina spike e positivo per le atre due porzioni del genoma virale che il test è in grado di rilevare.
Una seconda variante è stata osservata per la prima volta in campioni prelevati nel mese di ottobre in Sudafrica, dove è diventata la forma dominante del virus nel mese di dicembre. Questa variante, designata come B.1.351 o 501.V2, presenta mutazioni potenzialmente significative, e secondo analisi preliminari sarebbe caratterizzata anch’essa da maggiore trasmissibilità. In essa ritroviamo la mutazione N501Y precedentemente descritta per la variante inglese e altre due mutazioni rilevanti che riguardano sempre il segmento RBD della proteina spike: la E484K e la K417N. Queste mutazioni sembrano in grado di inibire, in tutto o in parte, il legame degli anticorpi neutralizzanti con la proteina modificata, dal momento che essi sono in grado di riconoscere quella contenuta nel virus originario (i virologi lo chiamano virus wild type o selvaggio), sequenziato da un paziente di Wuhan all’inizio del 2020).
Una terza variante è stata recentemente individuata in Brasile e in Giappone, dove è stata sequenziata in quattro viaggiatori provenienti dal Brasile. Anche questa, individuata con la sigla B.1.1.28.1 o P.1, si ritiene possa essere caratterizzata da maggiore contagiosità. In questa variante ritroviamo alcune mutazioni comuni a una o a entrambe le altre varianti: una delezione nel gene ORF1b (in comune con la variante inglese) e soprattutto alcune mutazioni nella proteina spike: K417N e E484K (in comune con la variante sudafricana) e la N501Y, che ritroviamo in tutte e tre le varianti.
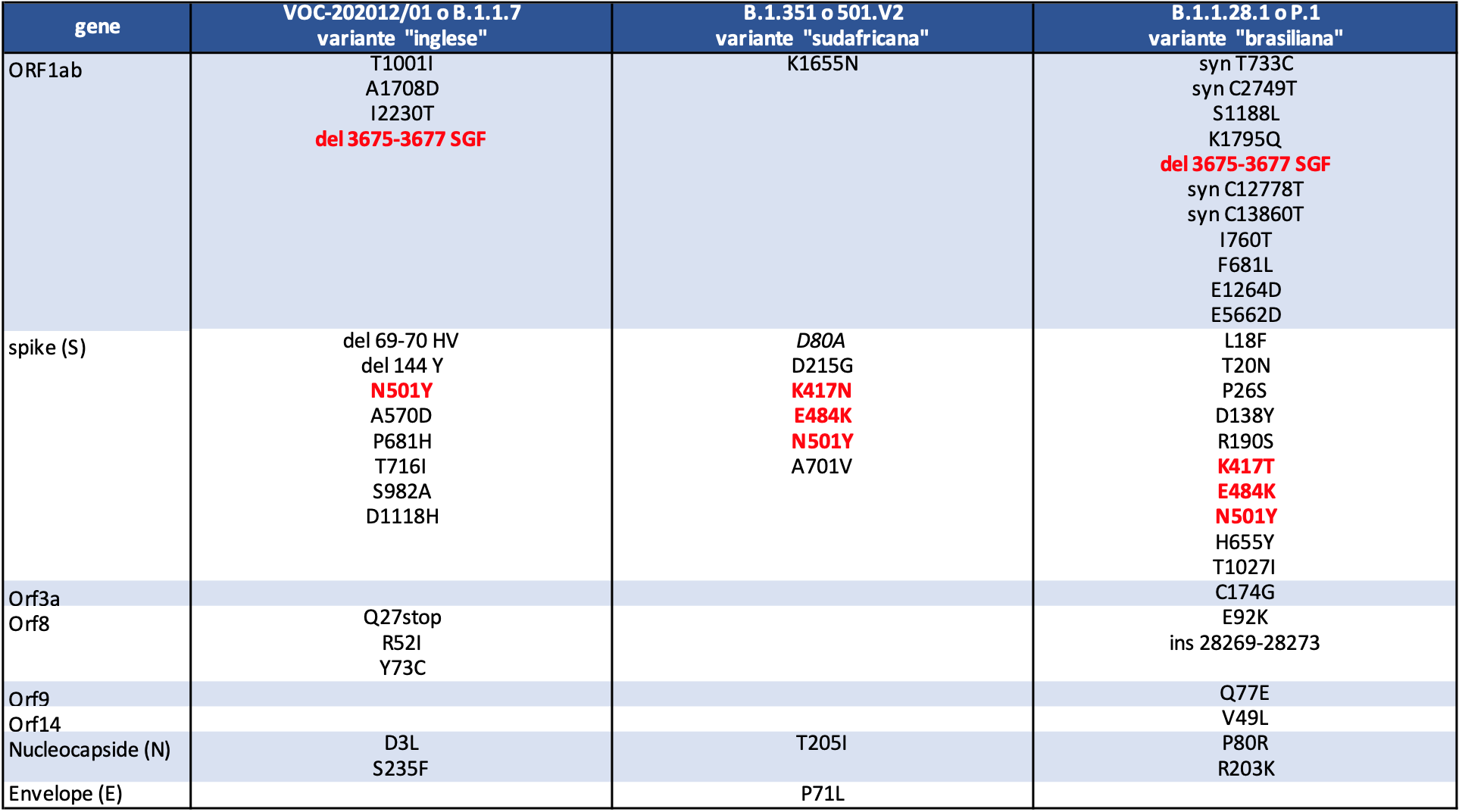
Figura 4. Variante inglese, sudafricana e brasiliana: le mutazioni. Legenda: syn, sinonimo; ins, inserimento; del, delezione; stop, traduzione interrotta. In rosso le mutazioni che ricorrono, totalmente o parzialmente, in più varianti.
Fonti: Galloway SE, Paul P, MacCannell DR et al. Emergence of SARS-CoV-2 B.1.1.7 Lineage — United States, December 29, 2020–January 12, 2021. MMWR Morb Mortal Wkly Rep . ePub: 15 January 2021; Rambaut A, Nick Loman et al. Preliminary genomic characterisation of an emergent SARS-CoV-2 lineage in the UK defined by a novel set of spike mutations, Dec 20, 2020, virological.org. Tegally E. Wilkinson et al. Emergence and rapid spread of a new severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) lineage with multiple spike mutations in South Africa, MedRxiv, Dec 22, 2020. Naveca F, Nascimento V et al. Phylogenetic relationship of SARS-CoV-2 sequences from Amazonas with emerging Brazilian variants harboring mutations E484K and N501Y in the Spike protein, Jan 11, 2021, virological.org. Faria NR, Morales Claro I et al. Genomic characterisation of an emergent SARS-CoV-2 lineage in Manaus: preliminary findings, Jan 13, 2021, virological.org
Cominciano intanto ad aumentare notizie dell’emersione di altre varianti: ultimamente abbiamo letto di una variante “californiana”, denominata L452R, che tra novembre e dicembre è salita dal 3,8% al 25% dei sequenziamenti effettuati in California, area dove l’incidenza del virus è in forte aumento. Un cluster di 35 persone positive a una versione “mutata” del virus è stato individuato nella stazione sciistica di Garmisch-Partenkirchen, sulle alpi bavaresi. È probabile che, aumentando l’attenzione anche mediatica sul tema e il numero di sequenziamenti virali, questi “avvistamenti” diventeranno sempre più numerosi, e gli scienziati avranno a disposizione quantità sempre maggiori di dati per studiare il fenomeno e cercare di disegnare con maggiore precisione la traiettoria evolutiva del virus.
La sorveglianza genomica
È importante sottolineare che alcune di queste mutazioni, per di più in aree “delicate” del virus come la proteina spike, sono emerse in modo indipendente in tutte e tre le varianti. Questo manifestarsi quasi simultaneo di mutazioni simili o identiche in campioni raccolti in paesi del mondo assai lontani tra loro suggerisce che si possa trattare di cambiamenti avvenuti per selezione naturale del SARS-CoV-2, sottoposto all’enorme pressione evolutiva derivante dal doversi adattare ad un animale, l’essere umano, con il quale sino a pochi mesi fa non aveva mai “convissuto”.
Se, come è lecito ipotizzare, queste mutazioni conferiscono al virus un vantaggio evolutivo, rendendolo per esempio più trasmissibile o meglio attrezzato per eludere una risposta immunitaria pre-esistente, ovvero determinata da precedenti infezioni o vaccinazioni, dobbiamo ragionevolmente attenderci un aumento della frequenza di varianti con queste caratteristiche. Tutti i virus, in maggiore o minore misura, hanno la capacità, detta viral escape, di mutare, eludendo in questo modo il sistema immunitario umano, continuando a riprodursi all’interno delle cellule e rendendo assai più complicato lo sviluppo di farmaci antivirali e di vaccini.
Alcuni ricercatori del MIT hanno recentemente creato tre modelli di viral escape per altrettante proteine virali, l’emoagglutinina del virus influenzale, la glicoproteina envelope dell’HIV e la proteina spike del SARS.CoV-2. Per creare questi modelli sono stati utilizzati algoritmi di machine learning originariamente sviluppati per il linguaggio naturale umano: in pratica, sono state considerate mutazioni rilevanti ai fini evolutivi quelle che preservano l'infettività virale ma danno al virus un aspetto diverso che lo rende non riconoscibile da parte del sistema immunitario, allo stesso modo in cui la sostituzione per errore di una o più parole in una frase ne preserva la correttezza grammaticale e sintattica ma può cambiarne completamente il significato.
Per cercare di capire quali sono le traiettorie evolutive del virus è necessario però effettuare il maggior numero possibile di sequenziamenti del genoma degli isolati virali. Attraverso questa attività è stato possibile, all’inizio dell’epidemia, identificare e “caratterizzare” rapidamente il virus, sviluppare e validare test diagnostici e avviare la ricerca sui vaccini. Ma oggi questa attività è ancora più importante di un anno fa, tant’è vero che l’Oms ha recentemente pubblicato10 una guida all’utilizzo del sequenziamento come strumento di salute pubblica.
L’incremento dei sequenziamenti è l’unico sistema che permetta di monitorare la diffusione di varianti virali in grado di “evadere” dai sistemi di difesa e cercare di comprendere l’evoluzione del virus: non è un caso se l’allarme sulla nuova variante sia giunto dalla Gran Bretagna, che sin da marzo 2020 ha deciso di investire in maniera massiccia sul sequenziamento del virus. Su oltre 380.000 sequenze virali depositate al 17 gennaio sulla banca dati internazionale Gisaid, oltre 165.000 sono state effettuate in Gran Bretagna, contro le circa 75.000 degli Stati Uniti. In termini di rapporto tra casi positivi e sequenziamenti la Gran Bretagna è seconda solo alla Danimarca, dove sono state effettuate tantissime sequenze, oltre 28.00011, anche a seguito della paura generatasi nel paese scandinavo qualche mese fa a seguito della diffusione dell’epidemia tra i visoni, allevati per la pelliccia soprattutto nell’area settentrionale del Paese. Gli animali, dopo aver contratto l’infezione dall’uomo, avevano a loro volta trasmesso il contagio ad altre persone, e al termine di questo tragitto di andata e ritorno si è osservata in molti isolati virali una mutazione (Y453F) nella proteina spike.
In Germania, dove sino a metà gennaio sono stati effettuati circa 3.500 sequenziamenti, il governo ha recentemente deciso di investire 200 milioni di euro nel potenziamento di questa attività e ha disposto l’obbligo per i laboratori di sequenziare almeno un campione su venti. In Italia i sequenziamenti effettuati al 17 gennaio sono poco più di 2.200, un numero che speriamo possa crescere rapidamente.
Varianti e vaccini
La prima cosa che emerge da tutte le analisi sin qui condotte è che queste nuove versioni del virus sono probabilmente più trasmissibili ma non più letali. Ciò ha un senso dal punto di vista evolutivo: la morte dell’ospite decreta infatti anche la morte del parassita, se non riesce a migrare verso un altro ospite, quindi la maggiore letalità non aumenta le chance di sopravvivenza del virus, non gli dà in effetti alcun vantaggio. Per il virus la situazione “ideale” è quella della coabitazione pacifica, come avviene per esempio con l’herpes zoster, il virus che causa la varicella e il fuoco di Sant’Antonio, che rimane annidato nei gangli nervosi, poco irrorati dal sangue e quindi difficilmente raggiungibili dagli anticorpi, e si risveglia solo quando il sistema immunitario è indebolito.
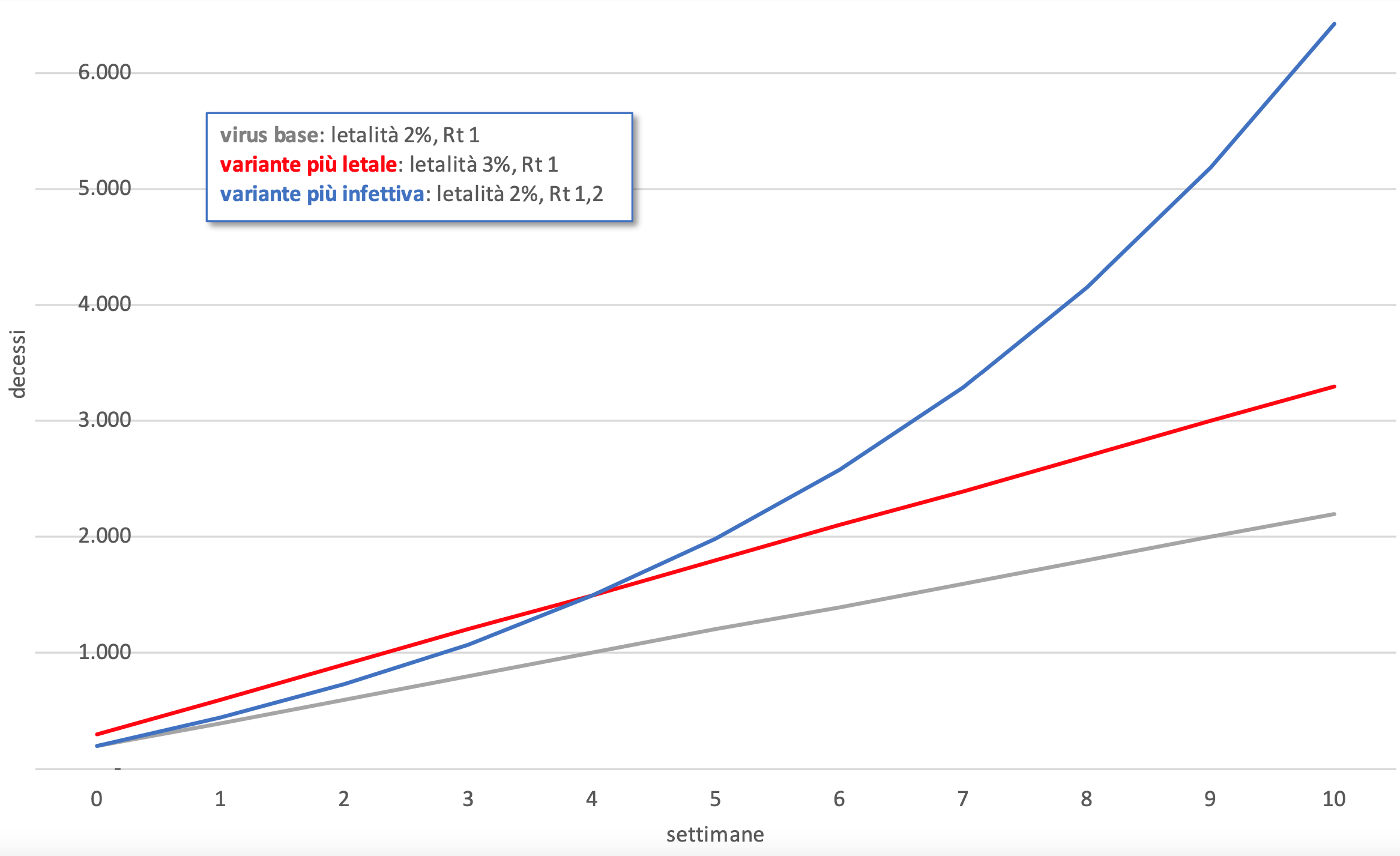
Figura 4. È peggio un virus più letale o uno più infettivo? Una simulazione dell’andamento dei decessi tra un virus “base”, un virus con uguale infettività ma più letale del 50%, e un virus con uguale letalità ma più infettivo del 20%
Tuttavia, il fatto che le varianti siano più trasmissibili ma non più letali, non deve essere motivo di grande consolazione. Immaginiamo una situazione di partenza con 10.000 contagiati, un tasso di letalità del 2%, quindi 200 decessi, e un numero di riproduzione Rt settimanale di 1, ovvero con il numero dei casi positivi stabile. In questa situazione, alla fine della settimana 10 avremmo un totale di 2.200 decessi. Se ipotizziamo di avere una variante virale più letale del 50% (quindi con tasso di letalità del 3%) alla fine della decima settimana i decessi sarebbero 3.300. Se invece la variante avesse la stessa letalità del virus originario ma una trasmissibilità maggiore soltanto del 20% (Rt 1,2), alla fine delle dieci settimane il numero complessivo degli infetti salirebbe notevolmente (quasi 62.000), e di conseguenza i decessi schizzerebbero a 6.430.
E dunque: mascherine, distanziamento, igiene delle mani. Le misure da applicare continuano a essere le stesse, e se aumenta la trasmissibilità del virus dovranno essere mantenute con ancora maggiore attenzione, in attesa di colpire il virus con la nostra prossima mossa, il vaccino.
Già, il vaccino. La domanda che tutti ci facciamo è: ci difenderà da queste e dalle altre varianti virali che emergeranno in futuro? La risposta immunitaria del vaccino, “tarata” sulla proteina spike del virus “selvaggio” isolato in tutto il mondo (Italia compresa) nel gennaio 2020, sarà efficace anche contro la proteina spike “mutata” contenuta nelle più recenti varianti virali? La risposta onesta a questa domanda è: non lo sappiamo ancora. I primi riscontri sembrano positivi: in uno studio per il momento pubblicato solo in preprint12, Pfizer e BioNTech hanno creato due pseudo-virus, uno con le caratteristiche del ceppo di Wuhan e l’altro con le mutazioni della variante inglese, e hanno riscontrato che gli anticorpi prodotti dal vaccino Pfizer hanno lo stesso effetto neutralizzante su entrambi. Altra buona notizia è che i vaccini contro il coronavirus, specialmente quelli realizzati con tecnologie di ingegneria genetica (come quelli a RNA messaggero o a vettore virale) sono facilmente adattabili ai nuovi ceppi virali. Una ragione in più, questa, per potenziare la sorveglianza genomica della pandemia e incrementare il numero dei sequenziamenti, con l’obiettivo di inserire nei futuri vaccini le sequenze “aggiornate” dei geni presenti nei ceppi virali dominanti. Qualcosa di simile a quanto avviene ogni anno con il vaccino anti-influenzale.
Dove andrà il virus?
Ci troviamo di fronte a una sorta di danza del cobra e della mangusta, dall’esito ancora incerto, tra il sistema immunitario umano, che reagisce a un patogeno di cui non ha traccia nella propria “libreria immunitaria” contenuta nei linfociti B e T, e un virus che cerca di adeguarsi ad un ambiente nuovo ed ostile. Non sappiamo ovviamente come evolverà la pandemia, ma se le attività di contenimento e di mitigazione dell’infezione non riusciranno a realizzare lo scenario di completa eradicazione di questo virus, uno degli scenari possibili è che esso diventi endemico. A oggi esistono, oltre al SARS-CoV-2, altri sei coronavirus in grado di trasmettersi da uomo a uomo. Due di questi, SARS CoV-1 e MERS, hanno tassi di letalità ancora più elevati del SARS-CoV-2, ma sono stati contenuti e non hanno mai avuto una diffusione sostenuta; gli altri quattro invece hanno una circolazione endemica, provocano sintomi lievi e non costituiscono una minaccia per la salute pubblica: causano infatti semplici raffreddori o manifestazioni para-influenzali.
L’analisi dei dati immunologici ed epidemiologici dei quattro coronavirus umani endemici evidenzia che, essendo virus molto diffusi, per questi patogeni la prima infezione avviene tra i tre e i cinque anni di vita, e prima dei quindici anni di età praticamente tutti siamo stati infettati. Nel prosieguo della nostra vita questi virus torneranno ripetutamente a visitarci, ma la prima infezione e le successive reinfezioni generano progressivamente una risposta immunitaria che, pur se non blocca del tutto le successive infezioni, ci protegge da manifestazioni cliniche di entità significativa.
Nel caso del SARS-CoV-2, siamo di fronte a un patogeno che non aveva mai avuto alcun contatto con l’essere umano e contro il quale dunque il nostro arsenale immunologico è sguarnito. II più giovani sembrano rispondere all’infezione da SARS-CoV-2 come alla prima infezione di uno dei quattro coronavirus endemici, con sintomi lievi o addirittura senza sintomi, mentre le persone di età più avanzata hanno più difficoltà. Con il tempo,e col progredire della vaccinazione della popolazione si potrebbe però costituire un substrato di immunizzazione: magari non perfetto, che si attenua col tempo, che non protegge del tutto dalle varianti, ma che comunque attenua i sintomi e li rende gestibili senza interessare le basse vie respiratorie e altri organi vitali. Non dunque una immunità di gregge che impedisca del tutto le infezioni, ma una “convivenza” con un virus le cui manifestazioni cliniche verrebbero a quel punto derubricate alla voce “malanno di stagione”.
Quanto tempo ci potrebbe volere? In una recente ricerca13, un gruppo di scienziati americani ha elaborato un modello matematico che, sulla base di vari parametri degli altri coronavirus umani endemici (età della prima infezione, tasso di riproduzione, durata dell’immunizzazione, severità della malattia nelle reinfezioni, tasso di letalità nella prima infezione), disegna una serie di scenari di passaggio da una fase di emergenza epidemica, nella quale il nuovo patogeno ha investito una popolazione completamente vergine causando una malattia severa nei gruppi più vulnerabili per età e patologie, a una fase endemica, nella quale gli individui si infettano da piccoli, poi continuano a reinfettarsi periodicamente e a trasmettere il virus. In questo scenario, la malattia Covid-19 si presenterebbe in forme progressivamente sempre più lievi, con tassi di letalità comparabili o anche inferiori a quelli dell’influenza stagionale, e la vaccinazione che stiamo avviando in questi mesi potrebbe giocare un ruolo fondamentale, accelerando nella popolazione, soprattutto in quella più anziana e fragile, l’acquisizione di una prima immunizzazione che altrimenti richiederebbe molto più tempo e soprattutto tanti decessi in più. Una maldicenza diffusa tra gli esperti dice che i modelli matematici, applicati alle malattie infettive, sono indubbiamente utili anche se non ci prendono mai. Speriamo invece che questa volta siano corretti.
Note
1. Brian Hie, Ellen D. Zhong, Bonnie Berger, Bryan Bryson, Learning the language of viral evolution and escape. Science, 15 Jan 2021 284-288. https://www.doi.org/10.1126/science.abd7331
2. van Dorp L, Richard, D, Tan CS et al. No evidence for increased transmissibility from recurrent mutations in SARS-CoV-2. Nat Commun 11, 5986 (2020). https://doi.org/10.1038/s41467-020-19818-2
3. Kupferschmidt K, U.K. variant puts spotlight on immunocompromised patients’ role in the COVID-19 pandemic. Science, 23 dicembre 2020. https://www.doi.org/ 10.1126/science.abg2911
4. Kemp SA, Collier DA et al. Neutralising antibodies drive Spike mediated SARS-CoV-2 evasion. MedRxiv, 19 dicembre 2020. https://doi.org/10.1101/2020.12.05.20241927
5. Choi B, Choudhary MC, Regan J et al. Persistence and Evolution of SARS-CoV-2 in an Immunocompromised Host. NEJM, 2020; 383:2291-2293 https://www.doi.org/10.1056/NEJMc2031364
6. Avanzato VA, Matson MJ et al. Case Study: Prolonged Infectious SARS-CoV-2 Shedding from an Asymptomatic Immunocompromised Individual with Cancer. Cell, Vol. 183, 7, P1901-1912.e9, December 23, 2020. https://doi.org/10.1016/j.cell.2020.10.049
7. WHO Disease Outbreak News, SARS-CoV-2 Variant – United Kingdom, 21 December 2020. https://bit.ly/38pm54p ECDC, Risk Assessment: Risk related to spread of new SARS-CoV-2 variants of concern in the EU/EEA, 29 dicembre 2020. https://bit.ly/3n7wkzL
8. Davies N, Barnard RC et al. Estimated transmissibility and severity of novel SARS-CoV-2 Variant of Concern 202012/01 in England, CMMID Repository, 23 dicembre 2020. https://bit.ly/37XAndf
9. Erik Volz1, Swapnil Mishra, et al., Report 42 - Transmission of SARS-CoV-2 Lineage B.1.1.7 in England: insights from linking epidemiological and genetic data. 31 dicembre 2020. https://bit.ly/3pFVVBb
10.Genomic sequencing of SARS-CoV-2: a guide to implementation for maximum impact on public health. Geneva: World Health Organization; 2021. https://bit.ly/3nxeYwc
11. European Centre for Disease Prevention and Control. Sequencing of SARS-CoV-2. 23 December 2020. ECDC:Stockholm; 2020 (https://www.ecdc.europa.eu/sites/default/files/documents/sequencing-of-S... )
12 Muik A, Wallisch AK et al. Neutralization of SARS-CoV-2 lineage B.1.1.7 pseudovirus by BNT162b2 vaccine-elicited human sera. BioRxiv, 19 gennaio 2021. https://doi.org/10.1101/2021.01.18.426984
13. Lavine JS, Bjornstad ON, Antia R, Immunological characteristics govern the transition of COVID-19 to endemicity. Science, 12 gennaio 2021, eabe6522. http://www.doi.org/10.1126/science.abe6522
